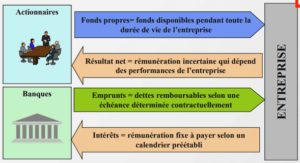
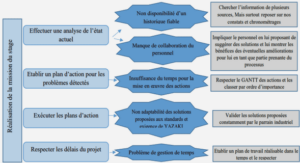
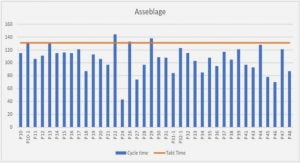
Avant de m'écrire sachez ceci :
1. La recherche est couteuse, donc il faut être prêt.e à investir dans votre réussite !
2. Vous êtes intéressé(e) par un document publié sur notre site ? merci de mentionner le lien de l'article sur la discussion !
** Demander un document (article ou livre), merci de mentionner le lien ou le titre sur la discussion !
3. Découvrez nos offres service complet : 1 service = 10$ min
4. Vous souhaitez publier un document dont vous êtes l’auteur sur notre site ?
Merci